

Abstract
Introduction: Daratumumab (Dara) is a human monoclonal antibody targeting the highly expressed multiple myeloma (MM) surface receptor CD38, with significant activity in relapsed MM. However, resistance to Dara develops in virtually all patients (pts). Thus, in order to maximize its clinical activity and prevent resistance, it is imperative to understand why pts stop responding. Our group recently reported that, in addition to the malignant cells, several immune-effector cells also express CD38. Thus, we hypothesized that the expression of CD38 or lack of it on non-cancer immune cell-subsets may also play a role in both clinical response and resistance to Dara.
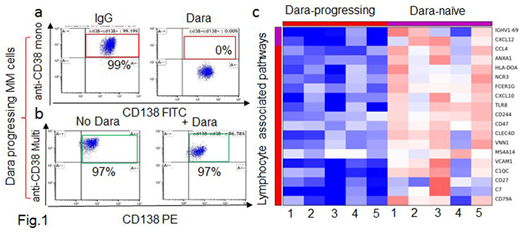
Results: To address our hypothesis, we analyzed CD38 surface expression in CD138+ MM cells isolated from 5 Dara-naïve (D-naïve) pts and from 8 Dara progressing pts (D-prg) who had discontinued Dara therapy for at least 4 weeks prior to analysis. We observed that the CD138+ MM-cells of D-prg displayed a lower median fluorescence intensity (MFI) of CD38 compared to the MFI of MM cells isolated from the D-naïve (p=0.04); no significant difference was however observed in the percentage of CD38+ MM cells between the two groups. In fact, all of the MM cells expressed CD38 in both D-naïve and D-prg, supporting the hypothesis that resistance to Dara was not mediated by loss of CD38 expression on MM cells. Next, we looked at whether CD38 on the surface of MM cells from D-prg was still recognizable by Dara. Thus, we treated MM cells ex-vivo with Dara at the clinically achievable concentration of 100 μg/ml. We showed that Dara was still able to bind CD38 on the surface of the D-prg CD138+ MM cells, since, after Dara treatment, no CD38+ signal was detected upon staining with a CD38 monoclonal antibody sharing the same targeting CD38 epitope (clone IB6) (Fig.1a). Conversely, we detected a CD38+ signal when a different anti-human CD38 multi-epitope antibody was used in a similar experiment (Cytognos) (Fig.1b). CD38 mRNA expression was observed in all MM cells isolated from D-prg, and we were not able to identify any missense mutations in CD38 mRNA from MM cells of D-prg for whom material was available for analysis.
Thus, having demonstrated that resistance to Dara was not related to the expression of CD38 on the MM-cells, we turned our attention to the MM bone marrow (BM) microenvironment (ME). We performed RNA-sequencing of the BM-ME depleted of the CD138+ MM cells obtained from the D-prg (n=5) and D-naïve (n=5). A gene expression signature differentiated the two groups. In D-prg, we observed a significant downregulation (fold-change<0.5, p<0.05) of 193 genes. DAVID functional annotation showed that the immune response-regulating genes, including TLR8, CD47, CXCL10 and CXCL4, were the most downregulated gene pathways in D-prg vs D-naïve (p<0.0001) (Fig.1c), supporting the notion that changes in the immune compartment of the ME may play a role in Dara resistance. Multi-parametric flow analysis showed that surface CD38 was significantly downregulated in terms of both MFI and percent in several immune cell subsets within the BM and peripheral blood (PB) of D-prg (n=14), in contrast to the BM and PB of the D-naïve (n=20) and the PB of the responding pts (n=6). Single cell RNA sequencing of PBMCs from naïve pts and D-prg is currently ongoing. Cell killing assays show that total PBMCs obtained from the D-prg, which have low/null CD38 expression in contrast to D-naïve and healthy donors, have no capability to kill CD38+ MM cells in the presence of Dara. However, total PBMCs obtained from D-naïve maintain an intact MM killing ability. Cell separation of the PBMC CD38+ and CD38- fractions obtained from healthy donors shows that Dara induced effector-mediated MM cell killing only in the CD38+ PBMCs treated with Dara; no killing effect was observed when the PBMCs CD38-negative fraction was used in the same experimental conditions. Moreover, killing assays show that Dara activates PBMCs to target CD38+ MM cells even when MM cells are not directly exposed to the antibody and only the effectors are treated.
Conclusions: Our data are supportive of the hypothesis that loss of Dara binding to MM-cell surface CD38 or the complete loss of CD38 on MM cells may not necessarily be the cause of Dara treatment resistance. We instead propose that Dara binding on CD38+ immune effector cells may be critical and that the loss of CD38 expression on these cells can lead to mechanisms of Dara-acquired resistance.
Rosenzweig:Celgene: Speakers Bureau. Krishnan:Celgene: Consultancy, Equity Ownership, Speakers Bureau; Janssen: Consultancy, Speakers Bureau; Sutro: Speakers Bureau; Takeda: Speakers Bureau; Onyx: Speakers Bureau.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal